
Machine Learning-Guided Design of Potent Darunavir Analogs Targeting HIV-1 Proteases: A Computational Approach for Antiretroviral Drug Discovery
Link: https://doi.org/10.1002/jcc.27298
Title: Machine Learning-Guided Design of Potent Darunavir Analogs Targeting HIV-1 Proteases: A Computational Approach for Antiretroviral Drug Discovery
Authors: Hathaichanok Chuntakaruk, Kajjana Boonpalit, Jiramet Kinchagawat, Fahsai Nakarin, Tanatorn Khotavivattana, Chanat Aonbangkhen, Yasuteru Shigeta, Kowit Hengphasatporn, Sarana Nutanong, Thanyada Rungrotmongkol, Supot Hannongbua
Abstract:
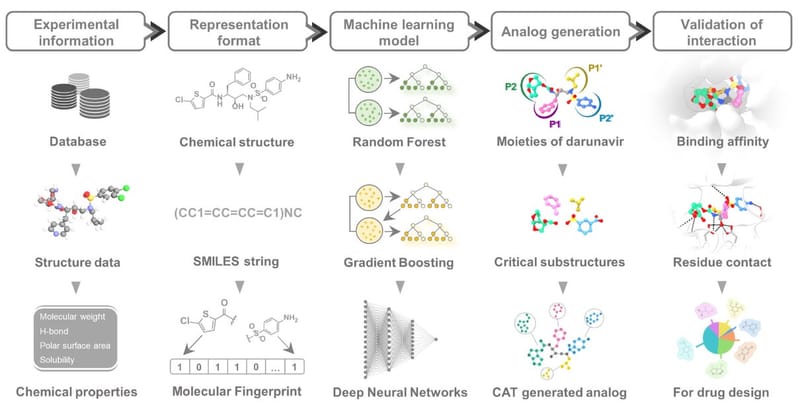
In the pursuit of novel antiretroviral therapies for human immunodeficiency virus type-1 (HIV-1) proteases (PRs), recent improvements in drug discovery have embraced machine learning (ML) techniques to guide the design process. This study employs ensemble learning models to identify crucial substructures as significant features for drug development. Using molecular docking techniques, a collection of 160 darunavir (DRV) analogs was designed based on these key substructures and subsequently screened using molecular docking techniques. Chemical structures with high fitness scores were selected, combined, and one-dimensional (1D) screening based on beyond Lipinski's rule of five (bRo5) and ADME (absorption, distribution, metabolism, and excretion) prediction implemented in the Combined Analog generator Tool (CAT) program. A total of 473 screened analogs were subjected to docking analysis through convolutional neural networks scoring function against both the wild-type (WT) and 12 major mutated PRs. DRV analogs with negative changes in binding free energy (ΔΔGbind) compared to DRV could be categorized into four attractive groups based on their interactions with the majority of vital PRs. The analysis of interaction profiles revealed that potent designed analogs, targeting both WT and mutant PRs, exhibited interactions with common key amino acid residues. This observation further confirms that the ML model-guided approach effectively identified the substructures that play a crucial role in potent analogs. It is expected to function as a powerful computational tool, offering valuable guidance in the identification of chemical substructures for synthesis and subsequent experimental testing.
Cite this:
Chuntakaruk, H.; Boonpalit, K.; Kinchagawat, J.; Nakarin, F.; Khotavivattana, T.; Aonbangkhen, C.; Shigeta, Y.; Hengphasatporn, K.; Nutanong, S.; Rungrotmongkol, T.; Hannongbua, S. Machine Learning-Guided Design of Potent Darunavir Analogs Targeting HIV-1 Proteases: A Computational Approach for Antiretroviral Drug Discovery. J. Comput. Chem. 2024, 45, 953-968. https://doi.org/10.1002/jcc.27298



